
Next-generation sequencing (NGS) is widely used for clinical diagnostics and to guide patient management. However, there are genomic regions difficult to analyze and variants difficult to detect using NGS. This is mainly because the sample sequence obtained from NGS must be aligned the human reference genome. After the alignment is completed any variants that differ from the reference are identified.
Limitations in variant detection
So far it all sounds simple and straightforward. So, what can go wrong?
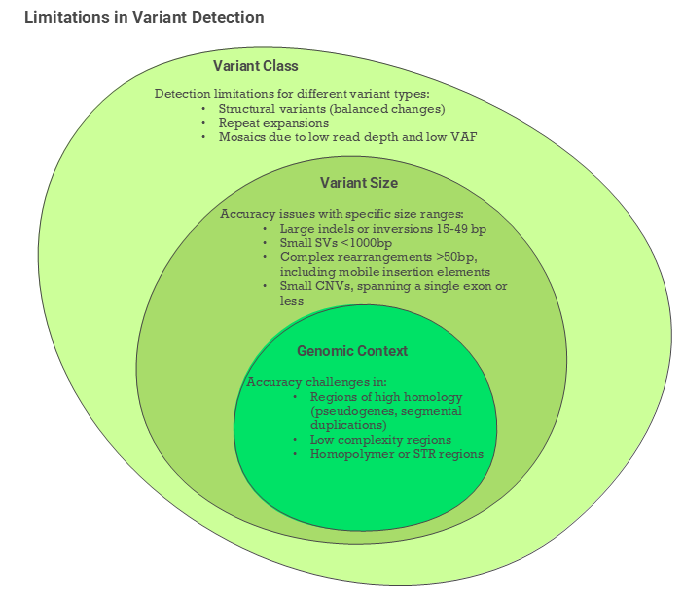
Not all regions are easy to align to each other. High-homology regions can match to multiple genomic locations making it difficult to determine where the variants belong. Examples include SMN1, GBA, CYP21A2, HBA1/HBA2, or PMS2. Tools used to filter out poor quality data can filter out low complexity regions (usually AT-rich) or homopolymer regions. This can lead to missing important variants in such regions. These can include for example CFTR intronic poly-T/poly-TG site, NPC1 c.1947+5G>C or MSH2 variant NM_000251.3:c.942+3A>T.
Most germline testing focuses on finding germline variants, present in about 50% of reads (in heterozygous state). This can miss mosaic variants present at much lower allelic frequency. Mosaic variants can be responsible for certain genetic conditions, so they are important to identify. Unfortunately, in WGS they can be removed by standard filtering.
One other highly important variant type that can be problematic in NGS are repeat expansions. Such expansions frequently cause neurological disorders. However, short-read NGS is not always capable of identifying them as some of the expansions are over 200 repeats, e.g., FGF14 deep intronic GAA expansion (over 300 GAA repeats). Long-read sequencing, such as PacBio or Oxford Nanopore can identify these expansions. Although some users still found GAA and AAG repeats challenging as they were filtered out by quality tools.
The long-read sequencing allows to improve some of the issues found with challenging variants. For example, long reads allow to resolve many of the structural variants, such as inversions, or translocations. We are also able to determine if variants are inherited together by phase analysis.
How do laboratories manage genetic testing?
Most of the laboratories use a variety of technologies for identification of genomic variants. Traditional methods are mostly inexpensive; however, they are mostly low throughput and have limited resolution. That said they are used in specific cases making them still a viable option for most clinical labs.
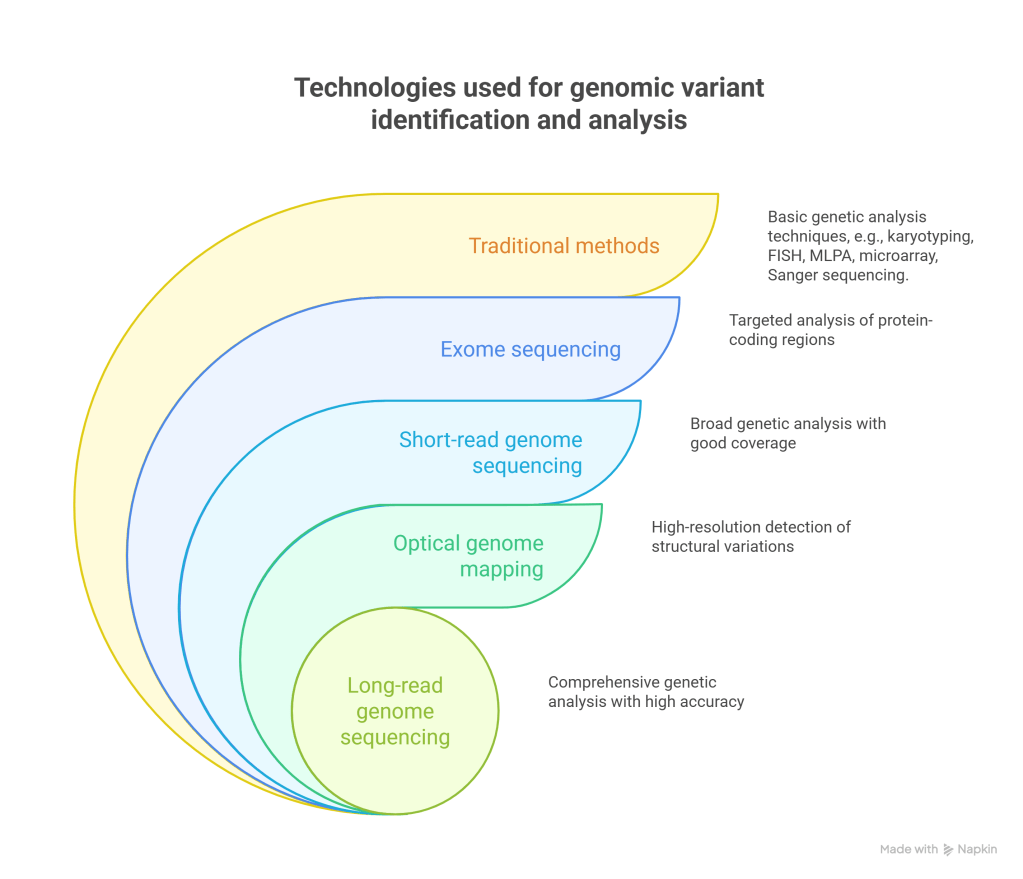
With the expansion of NGS into screening, workflows will need to be standardized to ensure similar performance across many laboratories. Software tools are evolving to include AI tools for data analysis. Yet, at the moment most of the workflows rely on a human review. As geneticists, we need to be confident that the tests have not missed an important variant. Whole genome sequencing (WGS) when applied at scale will put a lot of stress on the way labs analyze data.
We need to determine which variants or conditions cannot be tested reliably by WGS just yet. For such testing, the traditional methods will continue to be used.
How can we improve the test accuracy?
This is a complex issue. However, the laboratories use a variety of tools to develop and validate their tests. To be reimbursed, the laboratories also need to obtain approval or certification for their test. Once a test is approved, most of the labs are hesitant to introduce any changes to the test. They will still be working on improvements, but introduction of any changes is usually delayed till next re-certification round. If you are wondering why, making changes is not simple. In most cases partial or complete re-validation of the test is required or recommended to ensure performance is not affected.
During the test development the NGS workflow, including the chemistry needs to be optimized. The laboratories assess nucleic acid extraction, all the library prep components. They evaluate performance consistency of:
- Fragmentation and insert size
- Adapters used in the process
- Starting amounts of DNA and its quality. Certain sample types like blood spots, fixed tissues or plasma can affect the quality and quantity of DNA.
Wet lab workflow is just a part of the final WGS test. Bioinformatic analysis is the other critical part of a test. It plays two roles. One – to assess the performance of the assay, two – to identify variants in the samples. Therefore, chemistry and bioinformatics must be refined together.
Some of the difficult variants discussed above require specialized algorithms, for example:
- custom CNV caller.
- split-read detection algorithm – used for identifying precise breakpoints of inversions, translocations or large indels.
- specialized callers for homologous regions
- tools to accurately genotype variants embedded in repetitive sequences
Each laboratory must benchmark the test performance. It needs to establish the ranges for reporting, including targets, QC metrics, and so forth. It must also define the types and sizes of detectable variants. Additionally, any limitations must be established.
Reference materials for NGS
Ability to source a large number of clinical samples with specific genotypes can be problematic. There are exchange programs in the USA and Europe enabling clinical sample sharing between the labs for validation purposes. Additionally, the Genetic Testing Reference Materials Program (GeT-RM) coordinates studies to create well-characterized, publicly available genomic DNA reference materials for human genetic testing. Such materials are critical for development and validation of the new tests.
In general, there are three types of reference materials or controls:
- cell line-based reference materials
- mix of synthetic and native materials
- synthetic materials
There are some important needs such materials must fulfill. They must be method-agnostic and work in a variety of workflows. This is one of the requirements by the external quality assessment programs (EQAs). Bodies such as:
- GenQA (Genomic Quality Assessment),
- EMQN (European Molecular Genetics Quality Network),
- CAP (College of American Pathologists)
provide a variety of materials in their proficiency testing programs. The labs can have different tests, but still use the same material to participate.
Method-agnostic reference materials are important for another reason. Labs should be able to use them with orthogonal methods for confirmation.
What requirements do the reference materials or controls need to fulfill?
- Reference materials need to behave the same as the actual sample during the workflow. Commutability of the standards is very important as they need to be processed alongside the clinical samples. This way it controls every step of the laboratory workflow.
- Reference materials should include some of the challenging variants the laboratories are trying to identify.
- Ideally, the reference materials should contain a variety of variant types. Highly multiplexed standards can save time and money for the labs.
- The individual batches of materials must be consistent and should be produced to the highest standards of quality. Some of the ISO standards applicable are ISO 13485, ISO 17025, and ISO 17034.
- The data from the reference materials should be processed in the same way as the clinical samples. Many labs are using 3rd party software tools and have limited in-house bioinformatic support. Also, in the EQA programs, there is no provision for special processing of the controls.
Thus far, there is no requirement for the reference materials to be IVD or IVDR labeled. However, some manufacturers have obtained such certification. It is debatable if such labeling improves the quality of the materials beyond the ISO certification. It is important that all controls are manufactured according to ISO standards. This may be more important than IVD or IVDR labeling of the reference materials.
There are very few WGS reference materials. Most commonly used materials are:
- Genome in a bottle (GIAB),
- GeT-RM samples (selected Coriell samples that have been characterized),
- Coriell samples carrying specific variant (a wide range of these is available),
- Multiple samples, such as duos or trios.
Summary
The idea for this piece came out my own work as Lab Director, NGS assay and reference materials developer/product manager. Working with the clinical labs to provide them with the best tools for their testing was always important. As a geneticist I find it amazing how much progress we made. At the same time, it is amazing how many of the old sequencing problems are still with us.
How does the future shape?
- Long-read sequencing allows identification of variants which were too difficult for short-read technologies.
- Sequencing chemistries evolve to deliver better performance.
- Improved bioinformatic workflows and algorithms help to identify complex variants or variants in challenging regions.
- The availability of commutable reference materials containing challenging variants for assay development, validation, and ongoing quality monitoring is critical.
- Ensuring the quality of sequencing and comparability between the data across different laboratories should be our focus.
The topic not discussed here is the variant interpretation, which is another essential part of WGS. However, you cannot precisely interpret variants that are not detectable or missed due to technical issues. Here, proficiency testing providers play a critical role, enabling labs to continually assess and improve their performance.
References:
- Rojahn et al. (2022) Scalable detection of technically challenging variants through modified next‐generation sequencing. Mol Genet Genomic Med.10:e2072, DOI: 10.1002/mgg3.2072
- Lincoln, S et al (2021) One in seven pathogenic variants can be challenging to detect by NGS: an analysis of 450,000 patients with implications for clinical sensitivity and genetic test implementation. Genetics in Medicine 23:1673–1680; https://doi.org/10.1038/s41436-021-01187-w
- Ng, H. et al. (2025) Identification of technically challenging variants: Whole-genome sequencing improves diagnostic yield in patients with high clinical suspicion of rare diseases. Human Genetics and Genomics Advances 6,100469, https://doi.org/10.1016/j.xhgg.2025.100469.
- Marshall, C et al (2020) Best practices for the analytical validation of clinical whole-genome sequencing intended for the diagnosis of germline disease | npj Genomic Medicine. Genomic Medicine 5:47 ; https://doi.org/10.1038/s41525-020-00154-9
- Galbo, P. et al (2025) A Comprehensive Guide to Achieving New York State Clinical Laboratory Evaluation Program Approval for Next-Generation Sequencing Assays. The Journal of Molecular Diagnostics, Vol. 27, No. 6
- Genetic Testing Reference Materials Coordination Program | Laboratory Quality | CDC, accessed 25 July 2025
- Coriell Institute DNA and RNA samples catalog, accessed 29 July 2025
- LGC Clinical Diagnostics Science for a Safer World, accessed 29 July 2025
- Kernohan, KD and Boycott, KM (2024) The expanding diagnostic toolbox for rare genetic diseases. Nature Reviews Genetics 25, pages 401–415
- Hue, S et al (2019) CCMG practice guideline: laboratory guidelines for next-generation sequencing. Journal of Medical Genetics;56:792–800

Leave a comment